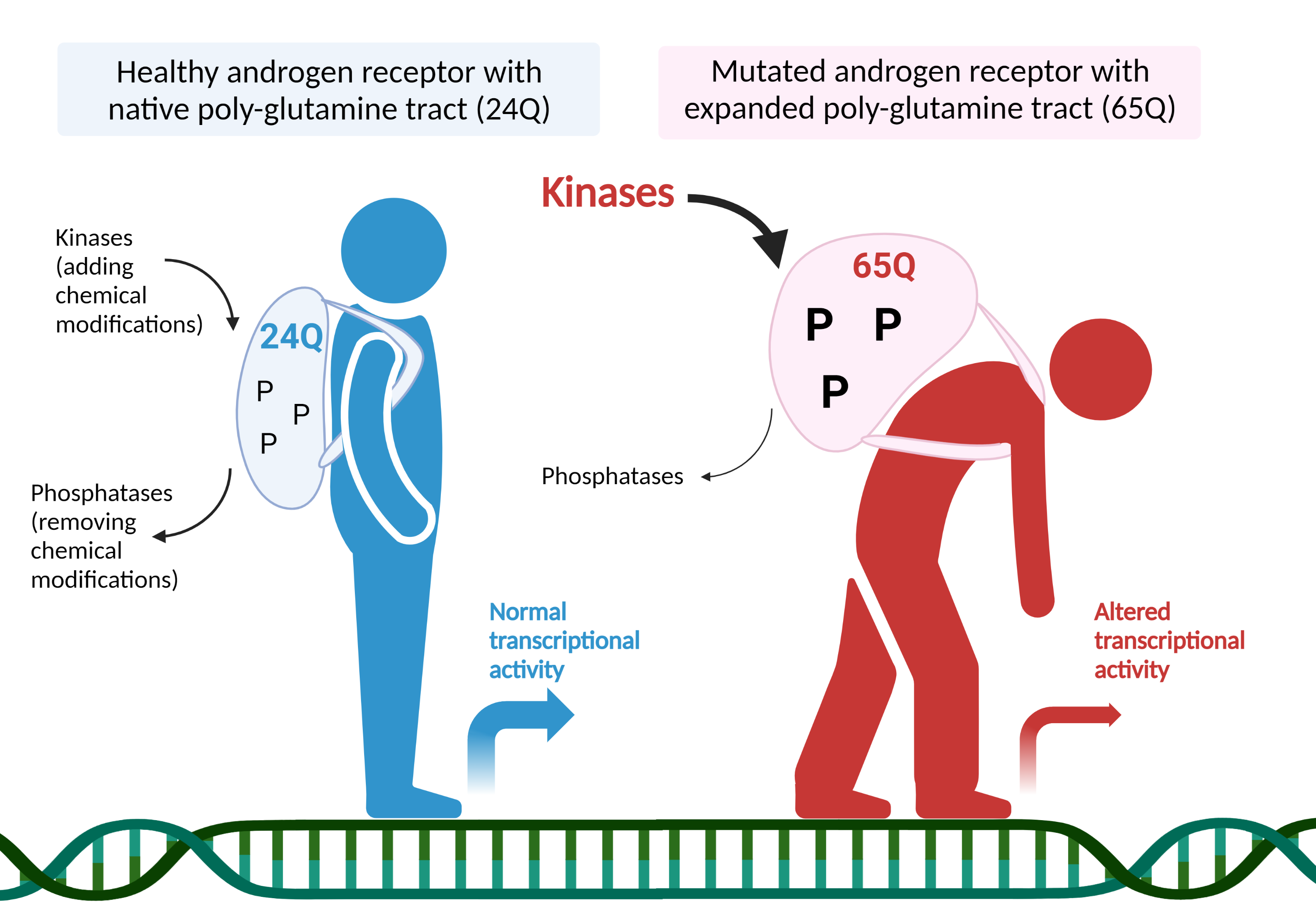


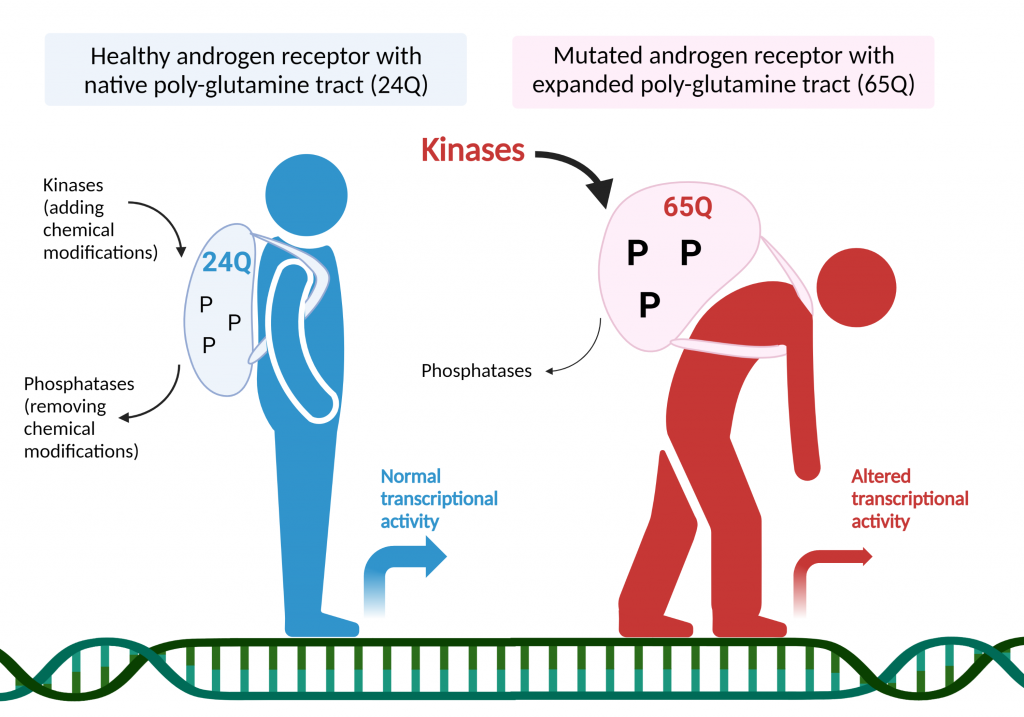
Spinal and bulbar muscular atrophy (SBMA) is a neurodegenerative disorder caused by the abnormal expansion of polyglutamine (PolyQ)-encoding CAG triplets in the androgen receptor (AR) gene. AR binds to testosterone and its more potent derivative dihydrotestosterone (DHT), which trigger the activation of androgen receptor and the transcription of genes responsive to testosterone. Several enzymes in the cells (called kinases) can add chemical modifications (phosphorylation) to transcription factors to tightly regulate specific gene expression programs. On the contrary, phosphatases do the opposite job and remove phosphorylation from these proteins. Working together in a concerted way, kinases and phosphatases modify stability, cellular localization and function of transcription factors. In this work, we revealed that the activation of cell division cycle 25 (CDC25)/cyclin-dependent kinase 2 (CDK2) pathway is a major contributor of mutant AR-dependent toxicity, in contrast to the activation of a calcium-dependent phosphatase, calcineurin (CaN), which mediates neuroprotection in vitro and in vivo, using motor neuron-like cell cultures, fly and mouse models. CDK2 was hyperactivated in the brainstem of diseased mice and its deletion reduced AR phosphorylation and restored the expression of genes involved in DNA damage, senescence and apoptosis, that were aberrantly expressed in SBMA mice. These observations highlight new disease modifiers and potential therapeutic approaches targeting kinases and phosphatases for the fine tuning of phosphorylation-mediated AR transcriptional repertoire that are aberrantly regulated in SBMA.
Piol D, Tosatto L, Zuccaro E, Anderson EN, Falconieri A, Polanco MJ, Marchioretti C, Lia F, White J, Bregolin E, Minervini G, Parodi S, Salvatella X, Arrigoni G, Ballabio A, La Spada AR, Tosatto SCE, Sambataro F, Medina DL, Pandey UB, Basso M, Pennuto M. Antagonistic effect of cyclin-dependent kinases and a calcium-dependent phosphatase on polyglutamine-expanded androgen receptor toxic gain of function. Sci Adv. 2023 Jan 6;9(1):eade1694. doi: 10.1126/sciadv.ade1694. Epub 2023 Jan 6. PMID: 36608116.